Pairwise and overall Fst with confidence intervals + building phylogenetic tree
Thierry Gosselin
2025-07-04
Source:vignettes/fst_confidence_intervals.Rmd
fst_confidence_intervals.RmdObjectives
- learn how to run the function
assigner::fst_WC84 - compute pairwise and overall Weir and Cockerham (1984) Fst estimates
- build confidence intervals
- visualize the estimates with data frame and matrices
- learn how to integrate the function with other package, e.g. to build a phylogenetic tree
- make Louis Bernatchez happy !
Special concerns for genome-wide estimate and filtering bias
During computation, the function first starts by keeping only the polymorphic markers in common between the populations.
When your project involves > 2 populations that evolved more by
neutral processes (e.g. genetic drift) than by natural selection,
filtering to keep only the markers in common between your populations
will drastically reduce the number of markers, and consequently your
genome coverage for the estimate. Strategically, you should reduce the
threshold of number of populations required to keep a marker (e.g. use a
minimum of 2 populations genotyped for the markers to keep it). This is
easily tested with radiator::filter_common_markers or in
stacks population module.
The overall fst and fis estimates will have reduce genome-wide coverage, because the computation is conducted with shared markers accross all your populations. However, for the pairwise comparisons, the markers will be tallied automatically for the pair. This usually results in more legitimate markers (i.e. less filtering bias) being used during computation, providing better genome-wide estimates for that pair.
Workflow
Preparation
- Clean your desk and remove the clutter in the global environment
- Required libraries
if (!require("pak")) install.packages("pak")
pak::pkg_install("thierrygosselin/assigner")
library(assigner)- Get the data:
data.fst <- readr::read_tsv(file = "https://raw.githubusercontent.com/thierrygosselin/package_data/master/assigner_data_fst.tsv.gz")Fst calculations
assigner::fst_WC84 first run: overall and pairwise, but
without boostrap (i.e. no confidence intervals). Get the function
documentation with: ??assigner::fst_WC84.
Results
After 10 sec max you should get the Fst (overall) value of 0.05
- To view the content of
fst:
names(fst)- Get access to specific content with
$:$sigma.loc: the variance components per locus:lsiga: among populations,lsigb: among individuals within populations,lsigw: within individuals.$fst.markers: the fst by markers$fst.ranked: the fst ranked$fst.overall: the mean fst overall markers and number of markers$fis.markers: the fis by markers$fis.overall: the mean fis overall markers and number of markers$fst.plot: the histogram of the overall Fst per markers$pairwise.fst: the pairwise fst in long/tidy data frame and number of markers per pairwise comparison$pairwise.fst.upper.matrix: the pairwise fst in a upper triangle matrix$pairwise.fst.full.matrix: the pairwise fst matrix (duplicated upper and lower triangle)$pairwise.fst.ci.matrix: matrix with pairwise fst in the upper triangle and the confidence intervals in the lower triangle.
- To view the pairwise Fst values in a tibble with 4 columns:
df <- fst$pairwise.fst-
The first 10 parwise comparisons in
dfN_MARKERS: is the number of markers used between the pairs to calculate the Fst.
POP1 POP2 FST N_MARKERS pop1 pop2 0.0442 3457 pop1 pop3 0.0507 3457 pop1 pop4 0.0341 3457 pop1 pop5 0.0300 3457 pop1 pop6 0.0434 3457 pop1 pop7 0.0367 3457 pop1 pop8 0.0256 3457 pop1 pop9 0.0475 3457 pop1 pop10 0.0522 3457 pop1 pop11 0.0473 3457 The matrix with fst values:
# to see as a tibble:
fst.matrix <- tibble::as_tibble(fst$pairwise.fst.full.matrix, rownames = "POP")
# to keep matrix:
fst.matrix <- fst$pairwise.fst.full.matrix- If you try to get the
pairwise.fst.ci.matrix:
- So let’s make another run with
fst_WC84, but this time will ask for the confidence intervals and the heatmap:
fst.ci <- fst_WC84(data = data.fst,
pop.levels = c("pop1", "pop2", "pop3", "pop4", "pop5", "pop6", "pop7", "pop8", "pop9", "pop10", "pop11"),
pairwise = TRUE,
ci = TRUE,
iteration.ci = 100,
quantiles.ci = c(0.025, 0.975),
parallel.core = 12,
heatmap.fst = TRUE,
filename = "testing_fst",
verbose = TRUE
)`fst.ci$pairwise.fst` is the tibble with the Fst values and now with the confidence intervals.- Try again to get the matrix with fst and ci:
fst.ci$pairwise.fst.ci.matrix- To have this in a handy table for publication, we will create a new
object
pairwise.fst.ci.df:
pairwise.fst.ci.df <- tibble::as_tibble(fst.ci$pairwise.fst.ci.matrix, rownames = "POP")
# if you have numeric pop_id identifier you might have to do this to get proper column names:
colnames(pairwise.fst.ci.df) <- colnames(fst.ci$pairwise.fst.ci.matrix)
# to save:
readr::write_tsv(x = pairwise.fst.ci.df, path = "pairwise.fst.ci.df.tsv")- Or even better, use the heatmap of Fst and CI values…:
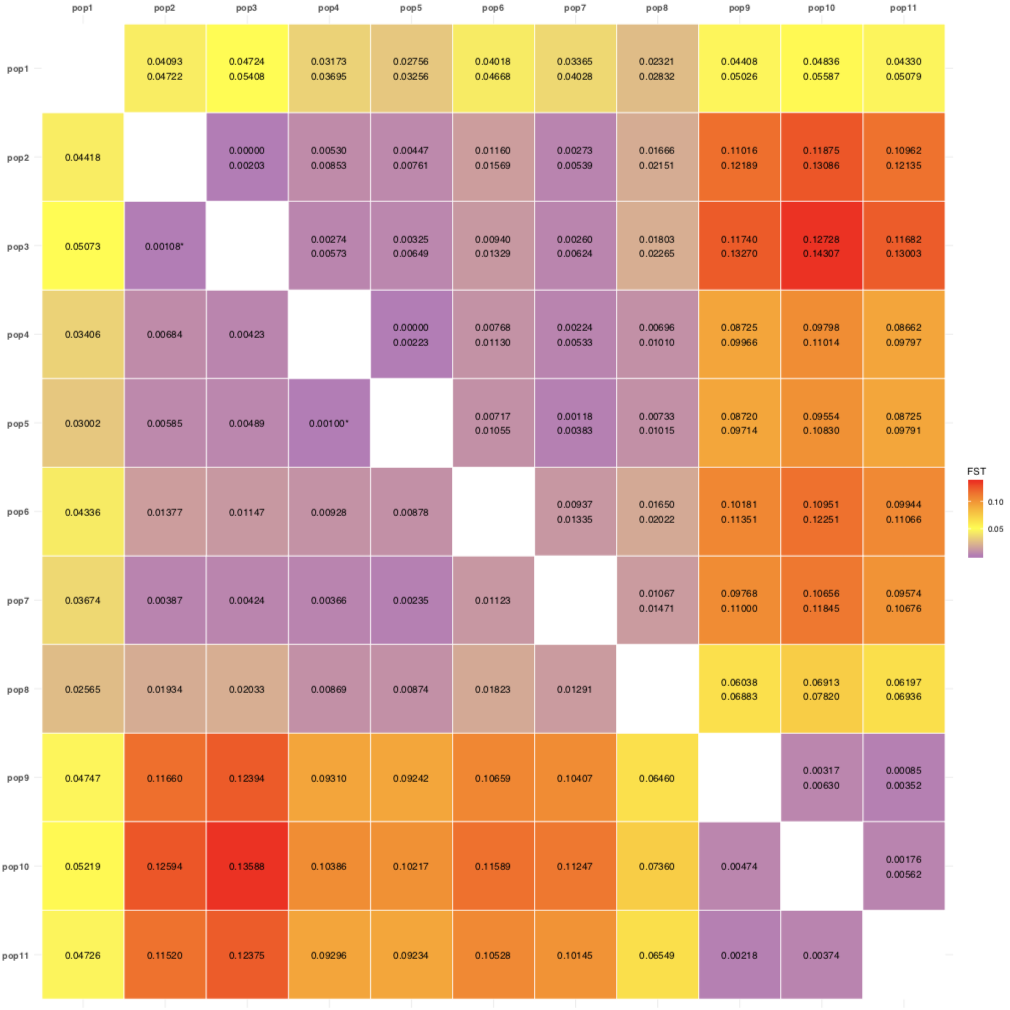
Phylogenetic tree
For the next steps, we need the full distance matrix created in step 10.
- For a Neighbor joining tree:
# build the tree:
require(ape)
tree <- ape::nj(X = fst.matrix) # fst.matrix as a matrix
# for annotating bootstraps values on the tree:
bootstrap.value <- ape::boot.phylo(
phy = tree,
x = fst.matrix,
FUN = function(x) ape::nj(x),
block = 1,
B = 10000,
trees = FALSE,
rooted = FALSE
)
# to get percentage values
bootstrap.value <- round((bootstrap.value/10000)*100, 0)
bootstrap.value
# to include in the tree
tree$node.label <- bootstrap.value - For a UPGMA tree:
require(stats)
tree <- ape::as.phylo(stats::hclust(stats::dist(fst.matrix), method = "average"))
bootstrap.value <- ape::boot.phylo(phy = tree, x = fst.matrix, FUN = function(xx) ape::as.phylo(stats::hclust(stats::dist(xx), method = "average")) , block = 1, B = 10000, trees = FALSE, rooted = TRUE)
bootstrap.value <- round((bootstrap.value/10000)*100, 0)
bootstrap.value
tree$node.label <- bootstrap.value- To build the tree we will need to install
# get the latest development version of ggtree:
if (!require("ggtree")) install_github("GuangchuangYu/ggtree")
# If it's not working, use the Bioconductor version:
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("ggtree")Several vignettes are available to get to know ggtree
Build a very basic tree figure:
require(ggtree)
require(ggplot2)
tree.figure <- ggplot2::ggplot(tree, ggplot2::aes(x, y), ladderize = TRUE) +
ggtree::geom_tree() +
# geom_tiplab(size = 3, hjust = -0.05, vjust = 0.5)+ # for just the tip label
ggplot2::geom_text(ggplot2::aes(label = label), size = 3, hjust = -0.05, vjust = 0.5) + # for both tips and nodes
ggtree::theme_tree() +
ggplot2::xlim(0, 0.16) # to allocate more space for tip labels (trial/error)
tree.figure
ggplot2::ggsave(filename = "assigner.fst.tree.example.pdf", width = 15, height = 15, dpi = 600, units = "cm", useDingbats = FALSE)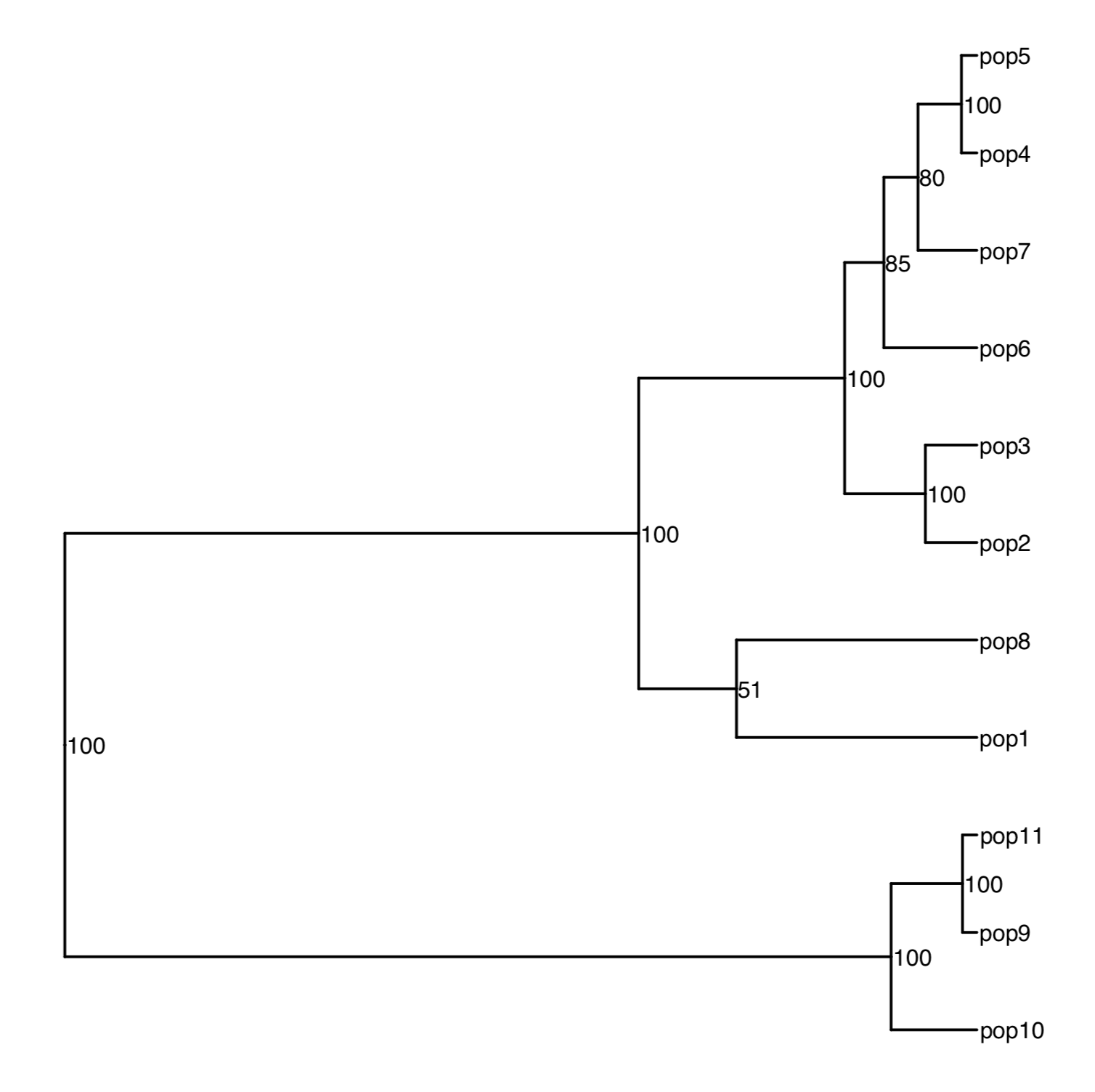
Conclusion
Please send me suggestions and bug reports through github
References
Weir BS, Cockerham CC (1984) Estimating F-Statistics for the Analysis of Population Structure. Evolution, 38, 1358–1370.
G Yu, D Smith, H Zhu, Y Guan, TTY Lam, ggtree: an R package for visualization and annotation of phylogenetic tree with different types of meta-data. revised.